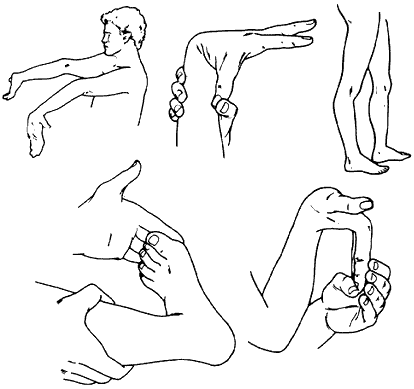
Ehlers-Danlos syndromes (EDS) is a hereditary disorder of connective tissue that cause a defect in collagen (a protein that acts as the body’s “glue” -adding strength and elasticity to connective tissue.) Collagen provides support to many body parts such as the skin, muscles, ligaments, and blood vessels. As a result of of faulty or reduced amounts of collagen, patients with EDS may have stretchy, fragile skin, fragile blood vessels prone to ruptures, and unstable joints.
EDS typically presents itself in young adulthood with hypermobile joints prone to misalignment, stretchy skin that tears easily, cardiovascular disease, and tissue fragility. There are 6 major types of EDS. The different types of EDS are classified according to their manifestations of signs and symptoms. Each type of EDS is defined as a distinct disorder that “runs true” in a family. This means that an individual with Vascular Type EDS may pass on Vascular Type EDS to their child, but not Classical Type EDS.
What are the signs and symptoms of EDS?
Joints
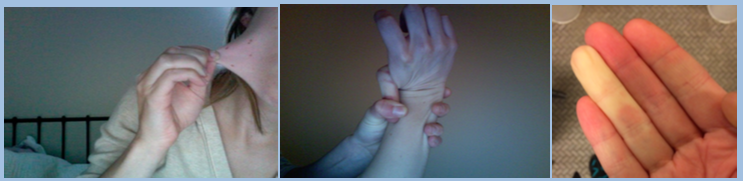
Joint hypermobility; loose/unstable joints which are prone to frequent dislocations and/or subluxations; joint pain; hyperextensible joints (they move beyond the joint’s normal range); early onset of osteoarthritis.
Skin
Soft velvety-like skin; variable skin hyper-extensibility; fragile skin that tears or bruises easily (bruising may be severe); severe scarring; slow and poor wound healing; development of molluscoid pseudo tumors (fleshy lesions associated with scars over pressure areas).
Cardiovascular
Mitral valve prolapse (heart valve abnormality), the tenancy to develop aneurisms, darterial/intestinal/uterine fragility or rupture (usually associated with the Vascular Type), autonomic dysfunction, raynaud’s phenomenon (loss of circulation to fingers and toes)
Miscellaneous/Less Common
Chronic, early onset, debilitating musculoskeletal pain (usually associated with the Hypermobility Type); scoliosis at birth and scleral fragility (associated with the Kyphoscoliosis Type); poor muscle tone (associated with the Arthrochalasia Type); Deformities of the spine, such as: scoliosis (curvature of the spine), kyphosis (a thoracic hump), tethered spinal cord syndrome, craniocervical instability, and Arnold–Chiari malformation (brain disorder).
Signs of EDS include: stretchy skin, hypermobile joints, and Reynolds phenomenon
How is EDS diagnosed?
Traditional diagnosis of EDS consists of family history and clinical evaluation to assess the diagnostic criteria. Most types of EDS can be diagnosed by skin biopsy. Genetic testing is also available for most types of EDS, although not for the most common type, Hypermobility. The tests vary in accuracy; in most instances genetic testing should be used conservatively to confirm an EDS type diagnosis rather than to rule one out.
How prevalent is EDS?
At this time, research statistics of EDS show the prevalence as 1 in 2,500 to 1 in 5,000 people. Recent clinical experience suggests EDS is more common. The condition is known to affect both males and females of all racial and ethnic backgrounds.
How is EDS inherited?
The two known inheritance patterns for EDS include autosomal dominant and autosomal recessive.
What is the prognosis of someone with EDS?
The prognosis depends on the type of EDS and the individual. While life-threatening complications can occur in all types of EDS, most people with EDS have a normal life expectancy unless they have the Vascular Type. There can be a wide or narrow range of severity within a family, but each person’s case of EDS will be unique. While there is no cure for EDS, there is treatment for symptoms, and there are preventative measures that are helpful for most.

{kind=link}