
Scientists will soon be developing a drug containing a potent and stable compound recently discovered within spider venom that blocks Nav1.7 channels. This discovery is a major breakthrough as previous research shows that people who have Congenital Insensitivity to Pain (CIP) lack Nav1.7 channels due to a naturally-occurring genetic mutation -so blocking these channels has the potential of turning off pain in people with normal pain pathways. Essentially, people who take this compound would develop an acquired form of CIP!
Individuals affected by CIP never feel pain in any part of their body when hurt. They can feel pressure, the difference between sharp and dull and hot and cold, but cannot sense pain, so they are likely to injure themselves without meaning to. Small children with this disorder often have to be bundled in protective gear so that they don’t hurt themselves as they haven’t yet learned what’s dangerous and what’s not. Eating feels like an unnecessary chore because they don’t feel hunger pains. Also, since the NaV1.7 channel is also found in olfactory sensory neurones, people with this condition usually have no sense of smell. But while a compound that induces this condition can be quite dangerous for some, it could also prove to be a life saver for people living with chronic pain.

People with CIP can feel heat, but cannot feel the pain of their feet burning.
As Professor King of the University of Queensland, an author of the study published in the British Journal of Pharmacology, said in the press release:
“Pain that cannot be controlled can ruin people’s lives. One in five people worldwide currently suffer from chronic pain, and existing pain treatments often fail to provide relief. The economic burden is huge, with chronic pain in the USA alone estimated to cost around $600 billion a year, greater than the combined economic cost of cancer, diabetes and stroke.”
Untapping this natural source of new medicine brings hope of accelerating the development of a new class of painkillers that can help people who suffer from chronic pain in a far more effective way than opioids and other analgesics. Moreover, since Nav1.7 channels are only involved in sensory neurons, there would likely be no side effects beyond loss of sensation, making it ideal for widespread pain conditions such as rheumatoid arthritis and fibromyalgia.
Sources:
http://www.eurekalert.org/pub_releases/2015-03/w-aos030315.php
http://ghr.nlm.nih.gov/condition/congenital-insensitivity-to-pain
http://en.wikipedia.org/wiki/Nav1.7#cite_note-SimpsonMcArthur2012-23
Journal Reference:
Julie K Klint, Jennifer J Smith, Irina Vetter, Darshani B Rupasinghe, Sing Yan Er, Sebastian Senff, Volker Herzig, Mehdi Mobli, Richard J Lewis, Frank Bosmans, Glenn F King. Seven novel modulators of the analgesic target NaV1.7 uncovered using a high-throughput venom-based discovery approach. British Journal of Pharmacology, 2015; DOI: 10.1111/bph.13081
WHAT IS EHLERS-DANLOS SYNDROME?
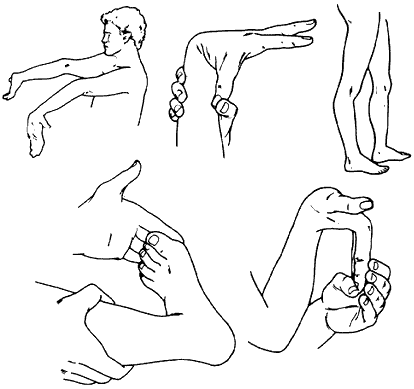
Ehlers-Danlos syndromes (EDS) is a hereditary disorder of connective tissue that cause a defect in collagen (a protein that acts as the body’s “glue” -adding strength and elasticity to connective tissue.) Collagen provides support to many body parts such as the skin, muscles, ligaments, and blood vessels. As a result of of faulty or reduced amounts of collagen, patients with EDS may have stretchy, fragile skin, fragile blood vessels prone to ruptures, and unstable joints.
EDS typically presents itself in young adulthood with hypermobile joints prone to misalignment, stretchy skin that tears easily, cardiovascular disease, and tissue fragility. There are 6 major types of EDS. The different types of EDS are classified according to their manifestations of signs and symptoms. Each type of EDS is defined as a distinct disorder that “runs true” in a family. This means that an individual with Vascular Type EDS may pass on Vascular Type EDS to their child, but not Classical Type EDS.
What are the signs and symptoms of EDS?
Joints
Joint hypermobility; loose/unstable joints which are prone to frequent dislocations and/or subluxations; joint pain; hyperextensible joints (they move beyond the joint’s normal range); early onset of osteoarthritis.
Skin
Soft velvety-like skin; variable skin hyper-extensibility; fragile skin that tears or bruises easily (bruising may be severe); severe scarring; slow and poor wound healing; development of molluscoid pseudo tumors (fleshy lesions associated with scars over pressure areas).
Cardiovascular
Mitral valve prolapse (heart valve abnormality), the tenancy to develop aneurisms, darterial/intestinal/uterine fragility or rupture (usually associated with the Vascular Type), autonomic dysfunction, raynaud’s phenomenon (loss of circulation to fingers and toes)
Miscellaneous/Less Common
Chronic, early onset, debilitating musculoskeletal pain (usually associated with the Hypermobility Type); scoliosis at birth and scleral fragility (associated with the Kyphoscoliosis Type); poor muscle tone (associated with the Arthrochalasia Type); Deformities of the spine, such as: scoliosis (curvature of the spine), kyphosis (a thoracic hump), tethered spinal cord syndrome, craniocervical instability, and Arnold–Chiari malformation (brain disorder).
Signs of EDS include: stretchy skin, hypermobile joints, and Reynolds phenomenon
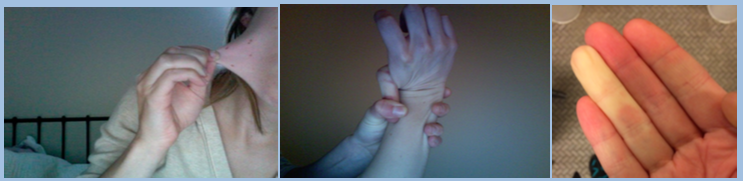
How is EDS diagnosed?
Traditional diagnosis of EDS consists of family history and clinical evaluation to assess the diagnostic criteria. Most types of EDS can be diagnosed by skin biopsy. Genetic testing is also available for most types of EDS, although not for the most common type, Hypermobility. The tests vary in accuracy; in most instances genetic testing should be used conservatively to confirm an EDS type diagnosis rather than to rule one out.
How prevalent is EDS?
At this time, research statistics of EDS show the prevalence as 1 in 2,500 to 1 in 5,000 people. Recent clinical experience suggests EDS is more common. The condition is known to affect both males and females of all racial and ethnic backgrounds.
How is EDS inherited?
The two known inheritance patterns for EDS include autosomal dominant and autosomal recessive.
What is the prognosis of someone with EDS?
The prognosis depends on the type of EDS and the individual. While life-threatening complications can occur in all types of EDS, most people with EDS have a normal life expectancy unless they have the Vascular Type. There can be a wide or narrow range of severity within a family, but each person’s case of EDS will be unique. While there is no cure for EDS, there is treatment for symptoms, and there are preventative measures that are helpful for most.
THE HUG THAT CHANGED MEDICINE

It’s not often that a hug changes the world, but for these two premature twins it did just that. In 1995 Brielle and Kyrie Jackson were born premature, and the doctors didn’t think they would survive until a nurse put them together.
When one of the twins took a turn for the worse, a nurse had the novel idea of taking the stronger twin and putting her in the same incubator as her sister -something that had never been done before in the U.S. The healthier sister then put her arm around the critically ill sister -her breathing and vital signs instantly stabilized.
Their hug is now famously known as “The Rescuing Hug” not only saved the twin’s life, but also changed the way that we take care of premature babies forever by highlighting the amazing healing power of touch.
The profound impact of skin to skin contact is known as Kangaroo care (named for the similarity to how certain marsupials closely carry their young.)
is a technique practiced on newborn, usually preterm, infants wherein the infant is held, skin-to-skin, usually by a parent. This ensures physiological and psychological warmth and bonding. Studies have found that skin-to-skin holding stabilizes heart and respiratory rates, improves oxygen saturation rates, better regulates an infant’s body temperature, and conserves a baby’s calories.
The video below catches us up with where they are today, and will make you grateful for their contribution to medicine:
WHAT IS CRANIOCERVICAL INSTABILITY?

Craniocervical Instability (CCI), also known as the Syndrome of Occipitoatlantialaxial Hypermobility, is a structural instability of the craniocervical junction which may lead to a pathological deformation of the brainstem, upper spinal cord, and cerebellum. It primarily occurs in patients with Ehlers-Danlos Syndrome and other hereditary disorders of connective tissue.
What is Ehlers-Danlos Syndome?
Ehlers-Danlos Syndome (EDS) is a genetic connective tissue disorder that is caused by a defect in the structure, production, or processing of collagen or proteins that interact with collagen. Collagen is kind of like your body’s glue -it is what holds your skin, joints, blood vessels, and other major organs in place. It typically presents itself in childhood or young adulthood with hypermobile joints prone to misalignment, stretchy skin that tears easily, and fragile blood vessels prone to cardiovascular complications (such as aneurysms).
EDS is currently considered to be a rare disease as it affects approximately 1 in 5000 people world wide (however, due to it’s vast underdiagnosis, a presumed frequency of 1 in 200 has been proposed).
EDS can be diagnosed by a skin biopsy, DNA blood testing, or clinically by a geneticist.
For more information on Ehlers-Danlos Syndrome click here.
How does Craniocervical Instability occur?
About 1 in 15 people with EDS will go on to develop CCI due to a lack of connective tissue support at the craniocervical junction. While some EDS patients present with this condition after a head and neck injury (such as whiplash), for the most part this condition tends to manifest gradually through repetitive stretch injuries from actions as simple as turning one’s head.
These stretch injuries can result in one or more (and in many times, all) of the following:
- Nerve dysfunction: deformative stress of repetitive stretching of the cranial-cervical nerves can lead to cell dysfunction and nerve death in this area.
- Thickened odontoid capsule (pannus formation): when joints are hypermobile, pannus may grow in a tumor-like fashion where it may erode articular cartilage and bone. When this occurs on the odointoid bone (a bone in our upper neck that acts as our head’s axis) it can compress the brainstem.
- Retroflexed Odontoid: Loose ligaments can misalign the proper angle of the odontoid bone causing it to push backwards, compressing the brainstem.
- Chiari Malformation: a downward displacement of the cerebellar tonsils (part of the brain) that puts pressure on the cerebellum and brainstem, progressively damaging them over time, and blocking the flow of cerebral spinal fluid (CSF).
- Cranial Settling: the skull sinking downward onto the spine. In severe cases basilar invagination occurs, where the tip of the odontoid process projects above the foramen magnum (the opening at the bottom of the skull).
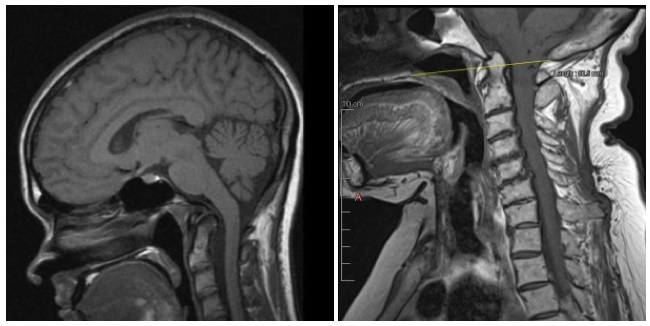
Unsurprisingly, the histopathological changes in neurons that are undergone in these situations (1) would not show up on any routine diagnostic test. In many cases, however, Chiari (4), cranial settling (5), and a retroflexed odontoid (3) may be demonstrable on MRIs, but typically only when imaged in the upright position. This would explain why many of these patients’ diagnostic imaging reports state negative results.
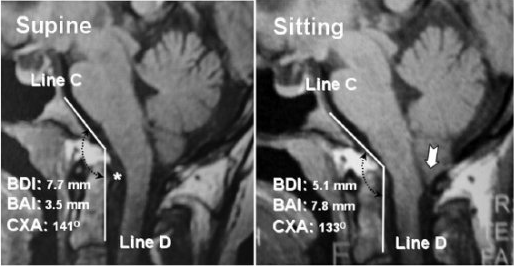
The following image shows a healthy looking MRI of an individual with EDS and CCI lying supine on the left compared with an MRI of the same individual in the upright position on the right. In the first image, the cerebellar part of the brain is neatly contained within the skull (i.e. where it should be) and the angle of the odontoid bone is within the normal limits. It is a radiographically normal MRI.
In the second image, however, there is a downward displacement of the cerebellar tonsils (demonstrated by downward arrow). The connective tissues here are too weak to hold up the cerebellum resulting in tonsillar herniation. Additionally, the odontoid bone is retroflexed, putting pressure on the brainstem.
What are the Symptoms of Craniocervical Instability?
- a heavy headache: a constant to near constant headache that can be described as feeling like the head is too heavy for the neck to support (feeling like a “bobble-head”)
- a pressure headache: an impairment of CSF flow causes intracranial pressure which would be aggravated by “valsalva maneuvers” such as yawning, laughing, crying, coughing, sneezing or straining.
- Dysautonomia: brainstem compression can lead to a dysfunctional autonomic nervous system (the involuntary regulator of all body functions). Symptoms of this include, but are not limited to:
- tachycardia (rapid heart)
- heat intolerance
- orthostatic intolerance (low blood pressure when standing)
- syncope (fainting)
- polydipsia (extreme thirst)
- delayed gastric emptying
- chronic fatigue
- Other symptoms include:
- neck pain
- central or mixed sleep apnea
- facial pain or numbness
- balance problems
- muscle weakness
- dizziness and vertigo
- vision problems
- reduced gag reflux and difficulty swallowing
- ringing in the ears and hearing loss
- nausea and vomiting
- impaired coordination
- downward nystagmus (irregular eye movements)
- paralysis
- and more
How is Craniocervical Instability Diagnosed:
Upright MRI and Rotational 3d CT scans are the standard imaging techniques used to determine if CCI is present individuals with EDS. A definitive diagnosis of CCI can be made by a technique known as Invasive Cervical Traction (ICT). ICT is an inpatient procedure where the paitent’s head is pulled upward by a pulley system. If, over the course of 48 hours, the patient’s symptoms are cleared, CCI is confirmed. Because ICT is rarely available in typical hospitals, as an alternative a doctor may simply pull the patient’s head up off the spine in the doctor’s office. If there is a reduction in pain and symptoms, it confirms the diagnosis. Patients may also have an extreme worsening of symptoms if their head is pushed downward.
What is the treatment for Craniocervical Instability?
Craniocervical fusion: a procedure in which the skull is pulled upward (cervical traction), placed into the corrected position, and then the occipital bone of the skull is fused to the upper cervical vertebrae to hold the corrected position. This procedure typically involves the use of rigid hardware, typically titanium (instrumented fusion) but can also involve a fusion construct (combination of bone, bone matrix, and sometimes bone morphogenic proteins) without hardware (noninstrumented fusion). Patients usually undergo rigid cervical immobilization following this procedure (cervical brace, halo vest, or custom minerva) until the occipital and cervical bones completely fuse together. In patients who also have a Chiari Malformation, the droopy and damaged cerebellar tonsils may be shrunk with electrocautery. This shrinkage ensures that there is no blockage of CSF flow out of the 4th ventricle.
References:
These Great Medical Conference Presentations:
WEBSITES:
http://csfinfo.org/education/physician-information/fusion-chiari-surgery/
ACADEMIC JOURNAL ARTICLES:
Milhorat, T. H., Bolognese, P. A., Nishikawa, M., McDonnell, N. B., & Francomano, C. A. Syndrome of occipitoatlantoaxial hypermobility, cranial settling, and chiari malformation type I in patients with hereditary disorders of connective tissue. Journal of Neurosurgery. 7, 601-609.
EHLERS-DANLOS SYNDROME HYPERMOBILITY TYPE: A GENETIC PREDISPOSITION TO THE DEVELOPMENT OF VARIOUS FUNCTIONAL SOMATIC SYNDROMES

Introduction
Functional Somatic Syndromes, conditions characterized by functional disability and self reported symptoms rather than clearly demonstrable organic problems, are a common contemporary health issue [1]. Each medical subspecialty seems to have at least one somatic syndrome for patients whose symptoms cannot otherwise be medically explained. These include: irritable bowel syndrome (gastroenterology); fibromyalgia (rheumatology); tension headaches (neurology); and chronic fatigue syndrome (immunology) [2]. In recent years, however, a significant portion of these patients have gone on to receive a diagnoses of a little known connective tissue disorder: Ehlers-Danlos syndrome hypermobility type (EDS-HT), formerly type III [3]. In this literature review, I will discuss the features of EDS-HT, explore EDS-HT as a possible unifying concept for various functional somatic syndromes, illuminate further implications of the described findings, outline a set of diagnostic criteria that should be implemented by healthcare professionals in functional diagnostic medicine, and propose a novel way of thinking about functional somatic syndromes.
This article was taken from https://chronically-awesome.com/eds-functional-somatic-syndromes/
EDS-HT, considered to be one and the same with joint hypermobility syndrome (JHS), is a relatively common, frequently underdiagnosed heritable condition which predisposes those afflicted to chronic, widespread musculoskeletal pain and a wide variety of articular and extra-articular features purportedly linked to constitutionally abnormal collagen. The diagnosis is primarily clinical in essence and is largely based on the Beighton score (a simple system used to quantify joint laxity and hypermobility) and medical history. It is predominantly of autosomal dominant inheritance, though the molecular basis of EDS-HT is still largely unknown except for a minority of patients mutated in TNXB and COL3A1 [4]. Skin biopsies may show alterations in collagen fibril morphology [5]. Early literature fixed the frequency of EDS as a whole to 1 in 5000, with EDS-HT accounting for approximately half of all registered cases. However, due to it’s vast underdiagnosis, a presumed frequency of 0.75-2% has been proposed for EDS-HT [4].
Hypermobility and the Autonomic Nervous System:
The Missing Link for Various Functional Somatic Syndromes
When first described, EDS-HT was considered to be a relatively benign condition, with acute and chronic joint instabilities as it’s unique clinical consequence. Recently, however, accumulated experience on the management of EDS-HT patients elucidated a more complex clinical picture. In particular, subjects with joint hypermobility appear to be more prone to developing a range of functional somatic syndromes [3], including fibromyalgia [6], chronic fatigue syndrome [7], headaches [8], complex regional pain syndrome [10], gastrointestinal functional disorder [11], pelvic organ prolapse [12], and orthostatic intolerance [13].
An underlying dysautonomic process may explain many of the aforementioned functional somatic syndromes seen in EDS-HT individuals, which are present in practically all major body systems. Leading research suggests that the pathogenic relationship between dysautonomia and congenital laxity of the connective tissue is primarily attributable to the pathological deformation of the brainstem and upper spinal cord from occipitoatlantoaxial hypermobility and cranial settling [8]. In other words, craniocervical hypermobility and instability, and the resulting deformative stress of repetitive stretching and ventral brainstem compression, appear to underlie the observed autonomic dysfunction in hypermobile patients [9]. As demonstrated in pathological reports of fatal cases of traumatic brain injury and numerous animal studies, repetitive stretching of nerves can lead to clumping and loss of neurofilaments and microtubules within the axon and promotes neural apoptosis [14][15]. Strain also alters the electro-chemistry of the nerve by decreasing the amplitude of action potentials [16] and increasing calcium influx into the cell [17]. When you apply this research to the context of hypermobile individuals, the underlying process of autonomic nervous system dysfunction becomes palpable. Unsurprisingly, the histopathological changes in neural axons that are undergone in these situations would not show up on any routine diagnostic test. In extreme cases, however, cranial settling and a reduction of the clivo-axial angle may be demonstrable on MRIs, but typically only when imaged in the upright position [8]. This would explain why many of these patients’ diagnostic imaging reports state negative results.
In accord with craniocervical hypermobility findings, recent studies have suggested that up to 70% of patients with hypermobility have orthostatic intolerance and other forms of dysautonomia. The orthostatic effect in EDS-HT individuals may also be compounded by abnormal connective tissue in the vasculature, which results in an increase in blood vessel distensibility in response to the augmented hydrostatic pressure that occurs during orthostatic stress. This leads to exaggerated blood pooling in the lower extremities with a resultant tachycardia [18]. While these findings were predictable, a reversed frequency study, wherein hypermobility was measured in patients diagnosed with Postural Orthostatic Tachycardia Syndrome, a prevalent form of dysautonomia in young people, found that an extraordinary 53% of participants met the diagnostic criteria for EDS-HT [19]. Furthermore, when hypermobility was measured in individuals diagnosed with Chronic Fatigue Syndrome, a condition with a longstanding, established association with orthostatic intolerance [20], researchers found that 25% of Chronic Fatigue syndrome sufferers had generalized hypermobility [21]. This phenomena, though, is likely of multifactorial consequence, as dysautonomia, chronic pain, and sleep apnea secondary to ventral brainstem compression can result in poor sleep architecture and chronic fatigue [22][23][24]. This article was taken from https://chronically-awesome.com/eds-functional-somatic-syndromes/
Ehlers-Danlos Syndrome Hypermobility Type as a Systemic Condition
The autonomic nervous system problems associated with hypermobility, alike various functional somatic disorders, are present in practically all major body systems. In the realm of gastroenterology, for instance, dysautonomia in the form of vagus nerve damage (which may result from craniocervical instability) can result in delayed gastric emptying [25] and affect bowel contractibility, causing nausea and the so called “irritable bowl syndrome” [26]. Moreover, the underlying collagen abnormality of EDS-HT itself is systemic. Insufficient collagen may reduce sphincter tone and increase distensibility of the gut wall (which is likely to influence the function of surrounding cellular mechano-receptors), resulting in decreased gastrointestinal motility, gastroesophageal reflux (GERD) and/or irritable bowel syndrome (IBS). In fact, over 50% of EDS-HT individuals have GERD and/or IBS [4][27]. When hypermobility was tested in patients diagnosed with functional gastrointestinal disorders (which include IBS, functional dyspepsia, and functional constipation), an astonishing 49% were found to have joint hypermobility and many of those patients went on to receive an official diagnosis of EDS-HT [10].
When it comes to neurological manifestations, headaches are among the most common complaint in the EDS-HT population [4]. As a consequence of occipitoatlantoaxial hypermobility, drooping of the cerebellar tonsils and obstruction of the cerebrospinal flow at the craniocervical junction can result in intracranial pressure [8][28]. In addition, rapid fluctuations in blood pressure and inadequate cerebral perfusion on upright posture caused by dysautonomia may lead to migraines [29][30]. People with lax joints are also predisposed to cerviocogenic, tension, and new daily persistent headaches arising from musculoskeletal dysfunction in the temporal mandibular joints and the upper three cervical segments of the spine [4][31].
As a consequence of ligamentous laxity, rheumatological complications among the EDS-HT population are commonplace. Chronic pain in patients with joint hypermobility stems from a predisposition to injury from daily minor trauma to the joints and ligaments [32]. Unstable joints may also lead to frequent dislocations, subluxations, sprains, and stretch injury to the nerves traversing hypermobile joints, further increasing the risk of developing chronic pain states such as arthralgia, repetitive strain injuries, and complex regional pain syndrome [4][9][33]. There is also a high incidence of muscular pain attributable to myofacial spasms. Tender points consistent with fibromyalgia are often palpable, especially in the paravertebral musculature [34]. In frequency studies, the prevalence of fibromyalgia in EDS-HT participants was established to be 30% [35] and the prevalence of EDS-HT among fibromyalgia subjects was found to be 27.3% [6]. One theory for the origin of pain in fibromyalgia ascribes it to excessive muscle stress, which may increase the excitability of nociceptive ends of the muscle [36][37]. Joint instability in hyperlax individuals may result in sustained muscle stress (an overcompensation mechanism for loose and injured joints) and over stimulation of nociceptive nerve endings (which are poorly supported by defective collagen fibrils) [38]. An alternative, although equivocal, theory has suggested that biomechanical disturbances in the cervical spine may play a role in the pathogenesis of fibromyalgia. In a controlled study of 161 cases of traumatic injury to the cervical spine (primarily “whiplash”), fibromyalgia was diagnosed in 21.6% of those with neck injury verses 1.7% control subjects with lower extremity fractures [39], bringing us back to the notion that craniocervical instability, and the subsequent neurological damage, may be the underlying process in the development of functional somatic syndromes.
Further Implications of Discussed Findings in the Diagnosis and Management of Functional Somatic Syndromes
These observations suggest that a careful examination for hypermobility and connective tissue abnormalities should be an integral part of functional diagnostic medicine. Pathological deformation of the brainstem and stretch injury to neural axons due to an underlying congenital ligamentous laxity, as discussed here in the case of EDS-HT, or acquired ligamentous instability, such as whiplash, may indeed be the missing link in the pathogenesis of various functional somatic syndromes.
In a literature review of functional somatic syndromes, Wessely and colleges concluded, “a substantial overlap exists between the individual syndromes and that the similarities between them outweigh the differences” and “patients with one syndrome frequently meet diagnostic criteria for another” [40]. For this subset of patients, generalized joint hypermobility may represent the common milieu for functional somatic syndromes with ubiquitous manifestations. The predispositions EDS-HT imposes would further explain why many of these patients are affected profoundly by emotional arousal (as it’s mediated by the autonomic nervous system) and muscle tension, and why patients with different syndromes share non-symptom characteristics such as sex (as joint laxity is more pronounced in females) and develops at a relatively young age (as EDS-HT is heritable, and hence, lifelong) [4][41].
Accordingly, articular hypermobility can be assessed by using the 9-point Beighton score, which assigns one point for each side of the body on which the patient can (1) passively dorsiflex the 5th finger >90 degrees with the forearm flat on the table, (2) passively appose the thumb to the flexor aspect of the forearm, (3) hyperextend the elbow beyond 10 degrees, and (4) hyperextend the knee beyond 10 degrees and one point for forward flexion of the trunk with the legs straight so that the palms rest flat on the floor. If a patient receives a Beighton score of 4 or more, a referral to a geneticist or rheumatologist for further evaluation is recommended [42]. If cranial settling and a reduction in the clivo-axial angle is suspected, and upright MRI may additionally aid in diagnosis [8].
With this hitherto unobserved connection comes a new line of treatment for a subdivision of patients with functional somatic disorders. Physical therapy, in the form of exercises that strengthen joint-supporting muscles, and bracing may provide joint stability and help minimize articular injury [4]. Elimination of brainstem deformation by straightening and stabilizing the craniocervical junction (via fusion surgery) may also improve pain, neural functioning, and quality of life [8].
Conclusion: A Paradigm Shift in the Etiology of Functional Somatic Syndromes
Disorders that lack “objective markers” are usually considered to be functional, not “organic.” This implies to some that the symptoms in functional somatic syndromes are physiological manifestations of psychosocial factors, a view that enforces an insular attitude to the etiology of disease rather than an interactive holistic approach. Consequently, when investigative results are negative, management is commonly limited to reassurance about the (apparent) absence of disease and occasionally psychiatric therapy. These treatments, however, are unpopular with patients, have low coherence rates, and seldom provide long-term therapeutic relief [41][43].
An alternative explanation is that the organic abnormalities are undetectable through cursory diagnostic testing as the underlying mechanism may be histopathological in origin, or, as seen in the case of upright MRIs on EDS-HT patients, the body may not be in the problematic position when testing takes place. The overly common cognitive error overshadowing high-tech medicine –that emotional issues are the underpinnings of illnesses lacking objectivity– must be overcome. While it is sufficient to say that, like virtually all known illnesses, psychosocial factors do play some role in functional somatic syndromes [1], an over emphasis on medically unexplained symptoms as being psychological bases causal reasoning on a negative. An absence of evidence does not denote an absence of organic disease –it simply means that the conditions that were tested for are not present in the individual and there is an infinite realm of alternative possibilities, such as EDS-HT.
Functional somatic disorders can only be successfully managed in the healthcare setting once a comprehensive understanding of their nature and treatment is acquired. The recognition of Ehlers-Danlos Syndrome Hypermobility type, and other disorders involving ligamentous laxity, as a possible physiological mechanism underlying various medically unexplained symptoms will help bridge the gap in physicians’ minds between described physical complaints and apparent negative test results in a subset of patients. Henceforth, in the wake of this disclosed correlation, further investigation into the role hypermobility and connective tissue abnormalities play in the etiology of these conditions, alongside a redefinition and modification of the diagnostic criteria of functional somatic syndromes, is essential to study of medically unexplained phenomena.
http://www.thepainrelieffoundation.com/conditions/eds-functional-somatic Disorders that lack “objective markers” are…
Posted by Tea Lynn Moore on Friday, April 22, 2016
References:
- Barsky, A. J., & Borus, J. F. (1999) Functional somatic syndromes. Annals of Internal Medicine, 130(11), 910.
- Wessely, S. & White, D. (2004) There is only on functional somatic syndrome. The British Journal of Psychiatry, 185(2), 95-96.
- Castori, M., Celletti, C., & Camerota, F. (2013) Ehlers-Danlos Hypermobility type: a possible unifying concept for various functional somatic syndromes. Rheumatology International. 33(3), 819-821.
- Castori, M. (2012) Ehlers-Danlos Syndrome, Hypermobility Type: An Underdiagnosed Hereditary Connective Tissue Disorder with Mucocutaneous, Articular, and Systemic Manifestations. ISRN Dermatology. 2012, 751768.
- Hausser, I., & Anton-Lamprecht, I. (1994) Differential ultrastructural aberrations of collagen fibrils in Ehlers-Danlos syndrome types I-IV as a means of diagnostics and classification. Human Genetics. 93(4), 394.
- Acasuso-Díaz, M., & Collantes-Estévez, E. (1998) Joint hypermobility in patients with Fibromyalgia syndrome. Arthritis Care and Research. 11(1), 39–42
- Castori, M., Celletti, C., Camerota, F., & Grammatico, P. (2011) Chronic fatigue syndrome is commonly diagnosed in patients with Ehlers- Danlos syndrome hypermobility type/joint hypermobility syndrome. Clinical and Experimental Rheumatology. 29(3), 597.
- Milhorat, T. H., Bolognese, P. A., Nishikawa, M., McDonnell, N. B., & Francomano, C. A. Syndrome of occipitoatlantoaxial hypermobility, cranial settling, and chiari malformation type I in patients with hereditary disorders of connective tissue. Journal of Neurosurgery. 7, 601-609.
- Henderson, F. C., Wilson, W. A., Mott, S., Mark, A., Schmidt, K., Berry, J. K., Vaccaro, A., Benzel, E. (2010) Deformative stress associated with an abnormal clivo-axial angle: A finite element analysis. Surgical Neurology International. 1(1) 30.
- Stoler, J. M., & Oaklander, A. L. (2006) Patients with Ehlers Danlos syndrome and CRPS: a possible association? 123(1), 204–209.
- Zarate, N., Farmer A.D., Grahame, R., Mohammed S. D., Knowles, C. H., Scott, S. M., & Aziz, Q. (2010) Unexplained gastrointestinal symptoms and joint hypermobility: is connective tissue the missing link? Neurogastroenterology and Motility. 22(3), 252.
- Aydeniz, A., Dikensoy, E., Cebesoy, B., Altindaf, O., Gursoy, S., Balat, O. (2010) The relation between genitourinary prolapse and joint hypermobility in Turkish women. Archives of Gynecology and Obstetrics. 281(2), 301–304.
- Rowe, P.C., Barron, D.F., Calkins, H., Maumenee, I.H., Tong, P.Y., Geraghty, M. T. (1999) Orthostatic intolerance and chronic fatigue syndrome associated with Ehlers-Danlos syndrome. The Journal of Pediatrics. 135(4), 494–499.
- Smith, D. H., Hicks, R., & Povlishock, J. (2013) Therapy development for diffuse axonal injury. Journal of Neurotrauma. 30(5), 307-323.
- Saatman, K. E., Abai, B., Grosvenor, C., Vorwerk, C. K., Smith, D.H., and & Meaney, D. F. (2003) Traumatic Axonal Injury Results in Biphasic Calpain Activation and Retrograde Transport Impairment in Mice. Journal of Cerebral Blood Flow & Metabolism. 23(1) 34-42.
- Guruprakash, D. K. (2011) Effects of strain and strain rate on axonal injury in a spinal nerve root model. Wayne State University Theses. Paper 74.
- Wolf, J. A., Stys, P. K., Lusardi, T., Meaney, D. & Smith, D. H. (2001) Traumatic Axonal Injury Induces Calcium Influx Modulated by Tetrodotoxin-Sensitive Sodium Channels. The Journal of Neuroscience. 21(6), 1923–1930.
- Grubb, B. P., Kanjwal, Y., & Kosinski, D. J. (2006) The Postural Tachycardia Syndrome: A Concise Guide to Diagnosis and Management. Journal of Cardiovascular Electrophysiology. 17(1), 108-112.
- Jiménez-Cohl, P., Earle, N. M., González, B. R., & Thieck, E. J. (2012) Postural orthostatic tachycardia syndrome (POTS): report of 15 cases. Revista Medica de Chile. 140(2), 145.
- Stewart, J. M, Gewitz, M. H., Weldon, A., Arlievsky, N., Li, K., & Munoz, J. (1999) Orthostatic intolerance in adolescent chronic fatigue syndrome. Journal of Pediatrics. 103(1), 116-121.
- Nijs, J., De Meirleir, K., & Truyen, S. (2004) Hypermobility in Patients with Chronic Fatigue Syndrome: Preliminary Observations. Journal of Musculoskeletal Pain. 12(1) 9-17.
- Bagai, K., Song, Y., Ling, J. F., Malow, B., Black, B. K., Biaggioni, I., Robertson, D., & Raj, S. R. (2011) Sleep disturbances and diminished quality of life in postural tachycardia syndrome. Journal of Clinical Sleep Medicine.7(2), 204–210.
- Smith, M. T., & Haythornthwaite, J. A. (2004) How do sleep disturbance and chronic pain inter-relate? Insights from the longitudinal and cognitive-behavioral clinical trials literature. Sleep Medicine Reviews. 8(2), 119-132.
- Howard, R. S., Henderson, F., Hirsch, N. P., Stevens, J. M., Kendall, B. E., Crockard, H. A. (1994) Respiratory abnormalities due to craniovertebral junction compression in rheumatoid disease. Annals of the Rheumatic Diseases. 53(2), 134-136.
- Feldman, M., Corbett, D.B., Ramsey, E.J., Walsh, J. H., Richardson, C. T. (1979) Abnormal gastric function in longstanding, insulin-dependent diabetic patients. 77(1), 12-17.
- Spaziani, R., Bayati, A., Redmond, K., Bajaj, H., Bienenstock, J., Collins, S. M. & Kamath, M. V. (2008), Vagal dysfunction in irritable bowel syndrome assessed by rectal distension and baroreceptor sensitivity. Neurogastroenterology & Motility. 20(4), 336–342.
- Gastroesophageal reflux and irritable bowel syndrome in classical and hypermobile EDS. Program Nr: 362 from the 1999 ASHG Annual Meeting H.P. Levy, W. Mayoral, K. Collier, T.L. Tio, C.A. Francomano.
- Sansur, C. A., Heiss, J. D., DeVroom, H. L., Eskioglu, E., Ennis, R., & Oldfield, E. H. (2003) Pathophysiology of headache associated with cough in patients with Chiari I malformation. Journal of Neurosurgery. 98(3), 453–458.
- Low, P. A., Sandroni, P., et al. (2009) Postural Orthostatic Tachycardia Syndrome (POTS). Journal of Cardiovascular Electrophysiology. 20(3), 352-358.
- May, A., & Goadsby, P. J. (1999) The trigeminovascular system in humans: pathophysiologic implications for primary headache syndromes of the neural influences on the cerebral circulation. Journal of Cerebral Blood Flow Metabolism. 19(2), 115-27
- Hall, T., Briffa, K., & Hopper, D. (2008) Clinical Evaluation of Cervicogenic Headache: A Clinical Perspective. Journal of Manual and Manipulative Therapy. 16(2), 73–80.
- Grahame, R., Hakim, A.J. (2008) Hypermobility. Current Opinion in Rheumatology. 20(1), 106-110.
- Pascarelli, E. F., & Hsu, Y. P. (2001) Understanding work-related upper extremity disorders: clinical findings in 485 computer users, musicians, and others. Journal of Occupational Rehabilitation. 11(1), 1–21.
- Levy, H.P. (2004) Ehlers-Danlos Syndrome, Hypermobility Type.
- Hudson, N., Starr, M. R., Esdaile, J. M., & Fitzcharles, M. A. (1995) Diagnostic associations with hypermobility in Rheumatology patients. British Journal of Rheumatology. 34(12), 1157-1161.
- Bennet, R. M. (1996) Fibromyalgia and the disability dilemma: a new era in understanding a complex, multidimensional pain syndrome. Arthritis and Rheumatism. 39(10), 1627-1634.
- Zimmermann, M. (1991) Pathophysiological mechanisms of fibromyalgia. The Clinical Journal of Pain. 7(1), 8-15.
- Child, A. H. (1986) Joint hypermobility syndrome: inherited disorder of collagen synthesis. Journal of Rheumatology. 13(2) 239-243.
- Buskila, D., Neumann, L., Vaisberg, G., Alkalay, D., Wolfe, F. (1997) Increased rates of fibromyalgia following cervical spine injury: a controlled study of 161 cases of traumatic injury. Arthritis and Rheumatism. 40(3), 446- 452.
- Wessely, S., Nimnuan, C., & Sharpe, M. (1999) Functional somatic syndromes: one or many? The Lancet.354(9182), 936–939.
- Allan, L., Escobar, J. et al. (2002) Psychosocial treatments for multiple unexplained symptoms: a review of the literature. Psychosomatic Medicine. 64(6), 939-950.
- Beighton P. (1993) McKusick’s heritable disorders of connective tissue: The Ehlers-Danlos syndromes. (5th ed.). St Louis: Mosby. 206.
- Donta, S. T., Clauw, D. J., Engel, C. C., et al (2003) Cognitive behaviour therapy and aerobic exercise for Gulf War veterans’ illnesses: a randomized controlled trial. Journal of the American Medical Association. 289(11), 1396 -1404
16 THINGS PEOPLE IN CHRONIC PAIN WANT YOU TO KNOW

1. We try really hard to look good

We often hear “But you don’t look sick!” but the truth is that most of us try very hard to pass as normal. We rest before going out and take our pain meds at the optimal time. At times we hurt so much and are tired from trying to play healthy that we feel like laying down right then and there, but we (usually) hold it in until we get home to our beds.

2. It’s not all in our heads

Just because you can’t see it, it doesn’t mean it isn’t there. Our pursuit of healthcare is not driven by hypochondria or need for attention, it’s driven by physical discomfort. What we are doing is looking for something to improve our quality of life, and sometimes find the cause of our pain if it is not known.
“Just because you can’t see it it doesn’t mean it isn’t there” CLICK TO TWEET

“Our pursuit of healthcare is not driven by hypochondria or need for attention, it’s driven by physical discomfort.” CLICK TO TWEET
3. We are not making a mountain out of a molehill.
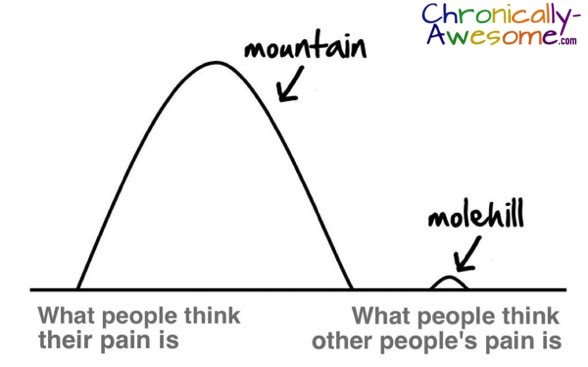
We are actually in more pain than you think we are in. Studies have shown that, generally speaking, people tend underestimate other people’s pain. This may be because chronic pain itself is difficult to imagine, especially if you have never experienced it firsthand. Even those who have experienced similar types of pain in the past have a difficult time remembering it until they experience it again.
4. No matter how long we’ve been suffering for, it still hurts

Having pain for an extended period of time does not give us superpowers to feel it less. However, most people with chronic pain have learned overtime to exhibit less pain related behaviours. So you can never really tell how much pain a person is in just by looking at them.
5. Sometimes we just don’t have the spoons

Spoon Theory is an analogy to explain what it’s like to live with a chronic illness such as chronic pain. Christine Miserandino, a woman who lives with lupus, originally coined the term on her website ButYouDontLookSick.com
The basic premise is that when you have a chronic condition you wake up each day with a certain number of spoons. Every time you exert effort — by getting out of bed, cleaning, getting dressed — you lose a spoon. When you run out of spoons, that’s it, the day’s activities are done. This article was written by https://www.chronically-awesome.com/16-things-chronic-pain/ -please follow link to view original.
Chronic pain can be an exhausting condition and this analogy demonstrates the need to budget and loss of control some people experience. So if we cancel our plans with you, it may be because we ran out of spoons.

6. We’re not lazy

In fact, we often have to work twice as hard to accomplish the tasks that most people do easily. This article is from https://www.chronically-awesome.com/16-things-chronic-pain/ –follow link to view original.

7. If we don’t have a job it’s for a reason

Some of us just don’t have the spoons to work on top of our activities of daily living. It can turn our pain from bearable to unbearable. Also, most employers are not eager to hire someone that can only work a few hours a week, is completely unreliable, may or may not show up, and may end up leaving at any point during the shift due to pain flares that make being productive impossible.
8. It’s really hard to get out of bed in the morning… and always!

But that doesn’t mean we still can’t have fun from bed.

So if we can’t make it out you can always bring the party to us!
9. Every minute feels like an eternity when waiting

Whether it’s an hour in a waiting room or 5 minutes in line, every minute drags out when you have to hold an uncomfortable position. It’s not that we are impatient, we would just prefer to use our spoons on more important things.
10. We are not ignoring you

Pain can be very distracting and mentally draining. We try our best to stay sharp and attentive but if we seem not to fully be there please don’t take it personally.

11. We get REALLY excited when we have a good day

Physically feeling good is just about the most exciting feeling in the world cause it means we can finally get stuff done! Its like going on a mini vacation (except for instead of doing nothing we try to do everything)!
12. And get really bummed when we have a bad day and can’t do the things we love

13. It can be hard to find a good doctor

Unfortunately, most health care professionals have little knowledge in pain management because it is rarely part of their training. We often go through many doctors before receiving a proper diagnosis and wait months to years (literally!) to see a pain specialist for treatment. Also, doctors too fall victim to the cognitive error of underestimating other’s pain, and vary few doctors are willing to take the legal risks involved in prescribing pain pills. So if we happen to find a good doctor who listens and is willing to treat us, we feel like we’ve died and gone to heaven! This article was written by https://www.chronically-awesome.com/16-things-chronic-pain/ -follow link to view original.
14. We are not drug seekers

We are pain relief seekers. Sometimes our medical treatment does require the use of opioids or medical marijuana to keep the pain under control and help us resume to as close to a normal life as we can. We take it just like any other medication. We dislike the side effects just like any other medication. And if we find pain relief from another means, we simply stop taking it, despite months or even years of use. This article was written by https://www.chronically-awesome.com/16-things-chronic-pain/ -please follow link to view original.
As the Cleveland Clinic explains: addiction appears to be distinctly uncommon in patients without a prior history of addiction. It’s important to keep in mind that addiction is different than physical dependence/tolerance. Physical dependence can occur with many different types of medications (e.g. beta-blockers), whereas addiction is a psychological phenomenon that is not caused by “chemical hooks” and usually requires a setting very different than that of a chronic pain patient. Unlike street-users, the medical patient is under the supervision of a doctor, is taking the medication in a slow-acting form, and is going home to a life where he or she is surrounded by the people they love.
“We are not drug seekers, we are pain relief seekers” CLICK TO TWEET
15. You don’t need to give us suggestions or medical advice

When we are venting about our situation what we really need is to be heard and understood, not given advice. We appreciate the thought, but it can be exhausting hearing advice all the time and frustrating when it doesn’t work. Unless we ask or you have chronic pain yourself, it’s best to leave this to the experts.
16. All we really need is your love and support

Sometimes all you can do is just be there, and that’s saving someone’s life!
read more articles by this author @ www.druthers.net
{kind=link}